Running a Wannier90 minimal workchain
GaAs wannierization workchain
This AiiDA workchain runs a full workflow from scratch (i.e., both Quantum ESPRESSO and Wannier90) to wannierise a GaAs crystal using manually-specified projections. The workflow returns the parsed information from the standard Wannier90 output (i.e. centres, spreads, etc.) and the interpolated band structure.
We use in the following the verdi command-line interface (CLI).
A reminder of a few useful AiiDA commands:
verdi process list -a -p <N>(to monitor the state of the calculations done in the last<N>days)verdi process show <identifier>(to list inputs and outputs of a calculation, especially useful after the calculation is finished)
Run your simulation
Open a terminal and enter the virtual environment where AiiDA is installed.
Then, enter the examples/workflows folder of the aiida-wannier90
plugin and run the following script to launch a new workflow:
./launch_w90_minimal.py -h
This will show the help of the command. You need to specify three
required parameters: a quantumespresso.pw code, a
quantumespresso.pw2wannier90 code and a wannier90.wannier90 code
(that you need to setup before). An example full command looks like:
./launch_w90_minimal.py -s pw@computer -p pw2wannier90@computer -w wannier90@computer
This will launch the simple workchain that is provided with
aiida-wannier90, whose class name is MinimalW90WorkChainand
that is found in the python module
aiida_wannier90.workflows.minimal.
Note: in this example we use sp3 orbitals centred on As atoms as starting guesses for projections. For more details about the inputs of QE and Wannier90 calculations you can inspect both the launcher and the workchain itself.
Check the results
When you run the launch_w90_minimal.py, on screen you will get an
output line for each step running (i.e. in order: pw.x SCF, pw.x
NSCF, wannier90.x preprocessing, pw2wannier.x, main
wannier90.x run), reporting the identifier (PK) of each calculation.
You can inspect each calculation node by typing, for instance:
verdi process show <PK>
to see the inputs and outputs of each calculation, or
verdi data dict show <PK>
to inspect the contect of Dict nodes, such as the
output_parameters of the Wannier90 calculation, containing the
parsed information from the output of the code.
As final results you get all the information parsed in AiiDA and saved as outputs, as documented in the previous sections.
Band structure
Once the calculation is finished, you export or visualize the resulting
bands interpolated using maximally-localised Wannier functions (MLWFs).
First, find the PK of the BandStructure returned by the workflow
using the command:
verdi node show <PK>
(passing the workflow PK). The BandStructure PK is found by looking
for the output node with link label wannier_bands.
With this BandStructure PK, you can show the bands with xmgrace
using:
verdi data bands show -F agr <PK>
or export it to file using:
verdi data bands export -F agr <PK> -o interpolated.agr
where interpolated.agr is the output file name where the band
structure will be stored.
Compare with DFT bands
You can compare the interpolated band structure with the bands obtained
directly from DFT (Quantum ESPRESSO). You can compute this band
structure, but for convenience we provide the DFT-bands.agr file in
the same folder. After having exported the MLWF-interpolated bands as
explained above, you can plot together both of them, executing the
following command:
xmgrace DFT-bands.agr interpolated.agr
You should see something like the following plot (where we just changed some graphical aspects):
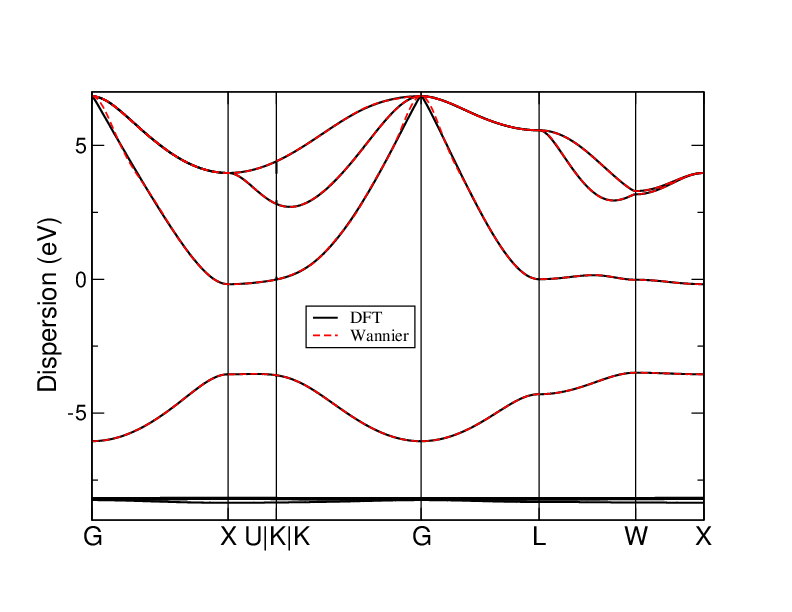
Comparison of DFT and Wannier band structures
Visualizing AiiDA provenance graphs
verdi node graph generate <PK>
where you can use the PK of a node for which you want to see the provenance (for instance a calculation, or the final band structure).
This command, by default, generates a PDF file named <PK>.dot.pdf,
that you can view with your favourite software.
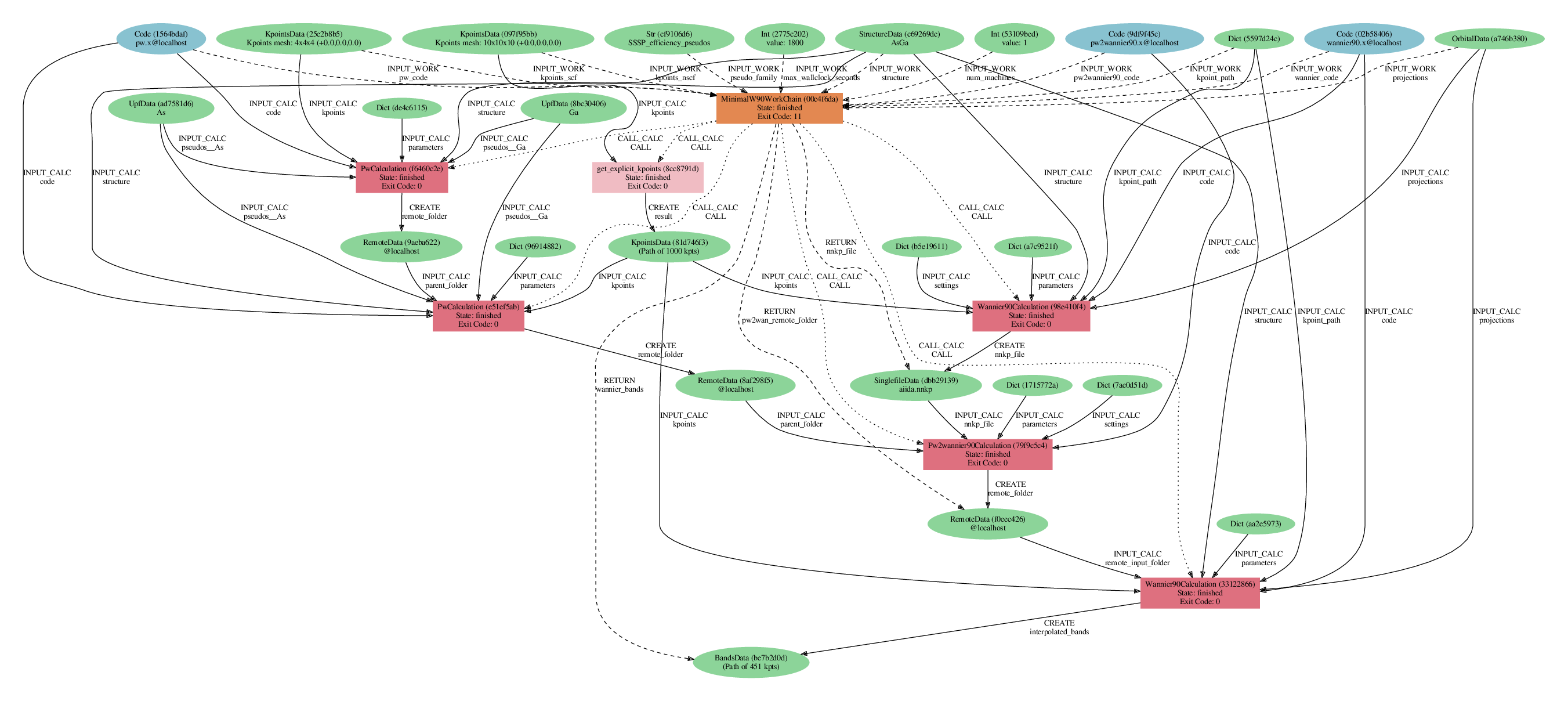
Example of the provenance graph of the band structure generated by the workflow